 |
REMICADE® (infliximab) is a chimeric IgG1k monoclonal antibody with an approximate molecular weight of 149,100 daltons. It is composed of human constant and murine variable regions. Infliximab binds specifically to human tumor necrosis factor alpha (TNF(alpha)) with an association constant of 10 10 M -1 . Infliximab is produced by a recombinant cell line cultured by continuous perfusion and is purified by a series of steps that includes measures to inactivate and remove viruses.
REMICADE is supplied as a sterile, white, lyophilized powder for intravenous infusion. Following reconstitution with 10 mL of Sterile Water for Injection, USP, the resulting pH is approximately 7.2. Each single-use vial contains 100 mg infliximab, 500 mg sucrose, 0.5 mg polysorbate 80, 2.2 mg monobasic sodium phosphate, monohydrate, and 6.1 mg dibasic sodium phosphate, dihydrate. No preservatives are present.
Infliximab neutralizes the biological activity of TNF(alpha) by binding with high affinity to the soluble and transmembrane forms of TNF(alpha) and inhibits binding of TNF(alpha) with its receptors. 1-4 Infliximab does not neutralize TNF(beta) (lymphotoxin (alpha)), a related cytokine that utilizes the same receptors as TNF(alpha). Biological activities attributed to TNF(alpha) include: induction of pro-inflammatory cytokines such as interleukins (IL) 1 and 6, enhancement of leukocyte migration by increasing endothelial layer permeability and expression of adhesion molecules by endothelial cells and leukocytes, activation of neutrophil and eosinophil functional activity, induction of acute phase reactants and other liver proteins, as well as tissue degrading enzymes produced by synoviocytes and/or chondrocytes. Cells expressing transmembrane TNF(alpha) bound by infliximab can be lysed in vitro by complement or effector cells. 2 Infliximab inhibits the functional activity of TNF(alpha) in a wide variety of in vitro bioassays utilizing human fibroblasts, endothelial cells, neutrophils, 3 B and T lymphocytes and epithelial cells. Anti-TNF(alpha) antibodies reduce disease activity in the cotton-top tamarin colitis model, and decrease synovitis and joint erosions in a murine model of collagen-induced arthritis. Infliximab prevents disease in transgenic mice that develop polyarthritis as a result of constitutive expression of human TNF(alpha), and, when administered after disease onset, allows eroded joints to heal.
Elevated concentrations of TNF(alpha) have been found in the joints of rheumatoid arthritis patients 5 and the stools of Crohn' disease patients 6 and correlate with elevated disease activity. In rheumatoid arthritis, treatment with REMICADE reduced infiltration of inflammatory cells into inflamed areas of the joint as well as expression of molecules mediating cellular adhesion [E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)], chemoattraction [IL-8 and monocyte chemotactic protein (MCP-1)] and tissue degradation [matrix metalloproteinase (MMP) 1 and 3]. 4 In Crohn' disease, treatment with REMICADE reduced infiltration of inflammatory cells and TNF(alpha) production in inflamed areas of the intestine, and reduced the proportion of mononuclear cells from the lamina propria able to express TNF(alpha) and interferon. 4 After treatment with REMICADE, patients with rheumatoid arthritis or Crohn' disease exhibited decreased levels of serum IL-6 and C-reactive protein (CRP) compared to baseline. Peripheral blood lymphocytes from REMICADE-treated patients showed no significant decrease in number or in proliferative responses to in vitro mitogenic stimulation when compared to cells from untreated patients.
Single intravenous infusions of 3 mg/kg to 20 mg/kg showed a predictable and linear relationship between the dose administered and the maximum serum concentration and area under the concentration-time curve. The volume of distribution at steady state was independent of dose and indicated that infliximab was distributed primarily within the vascular compartment. Median pharmacokinetic results for doses of 3 mg/kg to 10 mg/kg in rheumatoid arthritis and 5 mg/kg in Crohn' disease indicate that the terminal half-life of infliximab is 8.0 to 9.5 days.
Following an initial dose of REMICADE, repeated infusions at 2 and 6 weeks in fistulizing Crohn' disease and rheumatoid arthritis patients resulted in predictable concentration-time profiles following each treatment. No systemic accumulation of infliximab occurred upon continued repeated treatment with 3 mg/kg or 10 mg/kg at 4- or 8-week intervals in rheumatoid arthritis patients or patients with moderate or severe Crohn' disease retreated with 4 infusions of 10 mg/kg REMICADE at 8-week intervals. The proportion of patients with rheumatoid arthritis who had undetectable infliximab concentrations at 8 weeks following an infusion was approximately 25% for those receiving 3 mg/kg every 8 weeks, 15% for patients administered 3 mg/kg every 4 weeks, and 0% for patients receiving 10 mg/kg every 4 or 8 weeks. No major differences in clearance or volume of distribution were observed in patient subgroups defined by age or weight. It is not known if there are differences in clearance or volume of distribution between gender subgroups or in patients with marked impairment of hepatic or renal function.
The safety and efficacy of REMICADE when given in conjunction with methotrexate (MTX) were assessed in a multicenter, randomized, double-blind, placebo-controlled study of 428 patients with active rheumatoid arthritis despite treatment with MTX (the Anti-TNF Trial in Rheumatoid Arthritis with Concomitant Therapy or ATTRACT). Patients enrolled had a median age of 54 years, median disease duration of 8.4 years, median swollen and tender joint count of 20 and 31 respectively, and were on a median dose of 15 mg/wk of MTX. Patients received either placebo + MTX or one of 4 doses/schedules of REMICADE + MTX: 3 mg/kg or 10 mg/kg of REMICADE by intravenous infusion (IV) at weeks 0, 2 and 6 followed by additional infusions every 4 or 8 weeks in combination with MTX. Concurrent use of stable doses of folic acid, oral corticosteroids (</=10 mg/day) and/or nonsteroidal anti-inflammatory drugs was also permitted.
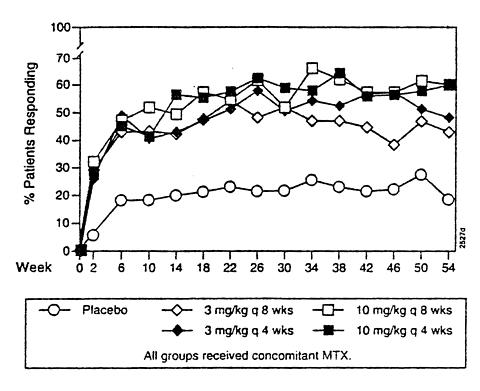
All doses/schedules of REMICADE + MTX resulted in improvement in signs and symptoms as measured by the American College of Rheumatology response criteria (ACR 20) 7 through 54 weeks (Figure 1).
|
|
Figure 1 Percentage of Patients who Achieved an ACR 20
Compared to placebo + MTX, all doses/schedules of REMICADE + MTX consistently resulted in greater effects on each component of the ACR 20, except for the HAQ, where only the 3 higher doses/schedules showed improvements in HAQ. Results from patients receiving 3 mg/kg q 8 weeks are shown in Table 1. Responses to the higher doses or more frequent administrations were similarly distributed.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
All doses/schedules of REMICADE + MTX resulted in a higher number of patients experiencing ACR 50 and ACR 70 compared to placebo + MTX (Table 2).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Health outcome measures were assessed by the SF-36 questionnaire. The eight subscales of the SF-36 were combined into two summary scales, the physical component summary (PCS) and the mental component summary (MCS). 9 At week 54, patients treated with 3 mg/kg or 10 mg/kg of REMICADE every 8 or 4 weeks showed significantly more improvement in the PCS compared to the placebo group, and no change in the MCS.
Radiographic Response
Structural damage in both hands and feet was assessed radiographically at week 54 by the change from baseline in the van der Heijde-modified Sharp score, a composite score of structural damage that measures the number and size of joint erosions and the degree of joint space narrowing in hands/wrists and feet. 10 Approximately 80% of patients had paired x-ray data. Results are shown in Table 3.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Data on use of REMICADE without concurrent MTX are limited (see Precautions, Immunogenicity ). 11,12
The safety and efficacy of REMICADE were assessed in a randomized, double-blind, placebo-controlled dose ranging study of 108 patients with moderate to severe active Crohn' disease 13 [Crohn's Disease Activity Index (CDAI) >/=220 and </=400]. All patients had experienced an inadequate response to prior conventional therapies, including corticosteroids (60% of patients), 5-aminosalicylates (5-ASA) (60%) and/or 6-mercaptopurine/azathioprine (6-MP/AZA) (37%). Concurrent use of stable dose regimens of corticosteroids, 5-ASA, 6-MP and/or AZA was permitted and 92% of patients continued to receive at least one of these medications.
The study was divided into three phases. In the first phase, patients were randomized to receive a single IV dose of placebo, 5, 10 or 20 mg/kg of REMICADE. The primary endpoint was the proportion of patients who experienced a clinical response, defined as a decrease in CDAI by >/=70 points from baseline at the 4-week evaluation and without an increase in Crohn' disease medications or surgery for Crohn' disease. Patients who responded at week 4 were followed to week 12. Secondary endpoints included the proportion of patients who were in clinical remission at week 4 (CDAI <150), and clinical response over time.
At week four, 4 of 25 (16%) of the placebo patients achieved a clinical response vs. 22 of 27 (82%) of the patients receiving 5 mg/kg REMICADE (p < 0.001, two-sided, Fisher' Exact test). One of 25 (4%) placebo patients and 13 of 27 (48%) patients receiving 5 mg/kg REMICADE achieved a CDAI <150 at week 4. The maximum response to any dose of REMICADE was observed within 2 to 4 weeks. The proportion of patients responding gradually diminished over the 12 weeks of the evaluation period. There was no evidence of a dose response; doses higher than 5 mg/kg did not result in a greater proportion of responders. Results are shown in Figure 3.
 |
Figure 3 Response (>/=70 point decrease in CDAI) to a Single IV REMICADE or Placebo Dose
During the 12-week period following infusion, patients treated with REMICADE compared to placebo demonstrated improvement in outcomes measured by the Inflammatory Bowel Disease Questionnaire.
In the second phase, 29 patients who did not respond to the single dose of 5, 10 or 20 mg/kg of REMICADE entered the open label phase and received a single 10 mg/kg dose of REMICADE 4 weeks after the initial dose. Ten of 29(34%) patients experienced a response 4 weeks after receiving the second dose.
Patients who remained in clinical response at week 8 during the first or second phase were eligible for the retreatment phase. Seventy-three patients were re-randomized at week 12 to receive 4 infusions of placebo or 10 mg/kg REMICADE at 8-week intervals (weeks 12, 20, 28, 36) and were followed to week 48. In the limited data set available, no significant differences were observed between the REMICADE and placebo re-treated groups.
The safety and efficacy of REMICADE were assessed in a randomized, double-blind, placebo-controlled study of 94 patients with fistulizing Crohn' disease with fistula(s) that were of at least 3 months duration. 14 Concurrent use of stable doses of corticosteroids, 5-ASA, antibiotics, MTX, 6-MP and/or AZA was permitted, and 83% of patients continued to receive at least one of these medications. Fifty-two (55%) had multiple cutaneously draining fistulas, 90% of patients had fistula(s) in the perianal area and 10% had abdominal fistula(s).
Patients received 3 doses of placebo, 5 or 10 mg/kg REMICADE at weeks 0, 2 and 6 and were followed up to 26 weeks. The primary endpoint was the proportion of patients who experienced a clinical response, defined as >/=50% reduction from baseline in the number of fistula(s) draining upon gentle compression, on at least two consecutive visits, without an increase in medication or surgery for Crohn' disease.
Eight of 31 (26%) patients in the placebo arm achieved a clinical response vs. 21 of the 31 (68%) patients in the 5 mg/kg REMICADE arm (p = 0.002, two-sided, Fisher' Exact test). Eighteen of 32 (56%) patients in the 10 mg/kg arm achieved a clinical response.
The median time to onset of response in the REMICADE-treated group was 2 weeks. The median duration of response was 12 weeks; after 22 weeks there was no difference between either dose of REMICADE and placebo in the proportion of patients in response (Figure 4). New fistula(s) developed in approximately 15% of both REMICADE- and placebo-treated patients.
 |
Figure 4 Response [fistula(s) closure] with Three Doses of REMICADE or Placebo
Seven of 60 (12%) evaluable REMICADE-treated patients, compared to 1 of 31 (3.5%) placebo-treated patients, developed an abscess in the area of fistulas between 8 and 16 weeks after the last infusion of REMICADE. Six of the REMICADE patients who developed an abscess had experienced a clinical response (see ADVERSE REACTIONS , Infections ).
Dose regimens other than dosing at weeks 0, 2 and 6 have not been studied. Studies have not been done to assess the effects of REMICADE on healing of the internal fistular canal, on closure of non-cutaneously draining fistulas (e.g., entero-entero), or on cutaneously draining fistulas in locations other than perianal and periabdominal.
REMICADE, in combination with methotrexate, is indicated for reducing signs and symptoms and inhibiting the progression of structural damage in patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to methotrexate.
REMICADE is indicated for the reduction in signs and symptoms of Crohn' disease in patients with moderately to severely active Crohn' disease who have had an inadequate response to conventional therapy.
The safety and efficacy of therapy continued beyond a single dose have not been established (see DOSAGE AND ADMINISTRATION ).
REMICADE is indicated for the reduction in the number of draining enterocutaneous fistulas in patients with fistulizing Crohn' disease.
The safety and efficacy of therapy continued beyond three doses have not been established (see DOSAGE AND ADMINSTRATION ).
REMICADE should not be administered to patients with known hypersensitivity to any murine proteins or other component of the product.
SERIOUS INFECTIONS, INCLUDING SEPSIS AND DISSEMINATED TUBERCULOSIS, HAVE BEEN REPORTED IN PATIENTS RECEIVING TNF-BLOCKING AGENTS, INCLUDING REMICADE. SOME OF THESE INFECTIONS HAVE BEEN FATAL. MANY OF THE SERIOUS INFECTIONS IN PATIENTS TREATED WITH REMICADE HAVE OCCURRED IN PATIENTS ON CONCOMITANT IMMUNOSUPPRESSIVE THERAPY THAT, IN ADDITION TO THEIR CROHN'S DISEASE OR RHEUMATOID ARTHRITIS, COULD PREDISPOSE THEM TO INFECTIONS.
CAUTION SHOULD BE EXERCISED WHEN CONSIDERING THE USE OF REMICADE IN PATIENTS WITH A CHRONIC INFECTION OR A HISTORY OF RECURRENT INFECTION. REMICADE SHOULD NOT BE GIVEN TO PATIENTS WITH A CLINICALLY IMPORTANT, ACTIVE INFECTION. PATIENTS SHOULD BE MONITORED FOR SIGNS AND SYMPTOMS OF INFECTION WHILE ON OR AFTER TREATMENT WITH REMICADE. NEW INFECTIONS SHOULD BE CLOSELY MONITORED. IF A PATIENT DEVELOPS A SERIOUS INFECTION INCLUDING SEPSIS, REMICADE THERAPY SHOULD BE DISCONTINUED (see ADVERSE REACTIONS , Infections ). PATIENTS SHOULD BE EVALUATED FOR THE RISK OF TUBERCULOSIS, INCLUDING LATENT TUBERCULOSIS. 15 TREATMENT FOR TUBERCULOSIS SHOULD BE INITIATED PRIOR TO TREATMENT WITH REMICADE.
REMICADE has been associated with hypersensitivity reactions that vary in their time of onset. Most hypersensitivity reactions, which include urticaria, dyspnea, and/or hypotension, have occurred during or within 2 hours of infliximab infusion. However, in some cases, serum sickness-like reactions have been observed in Crohn' disease patients 3 to 12 days after REMICADE therapy was reinstituted following an extended period without REMICADE treatment. Symptoms associated with these reactions include fever, rash, headache, sore throat, myalgias, polyarthralgias, hand and facial edema and/or dysphagia. These reactions were associated with marked increase in antibodies to infliximab, loss of detectable serum concentrations of REMICADE, and possible loss of drug efficacy. REMICADE should be discontinued for severe reactions. Medications for the treatment of hypersensitivity reactions (e.g., acetaminophen, antihistamines, corticosteroids and/or epinephrine) should be available for immediate use in the event of a reaction (see ADVERSE REACTIONS , Infusion-related Reactions ).
Infliximab and other agents that inhibit TNF have been associated in rare cases with exacerbation of clinical symptoms and/or radiographic evidence of de-myelinating disease. Prescribers should exercise caution in considering the use of REMICADE in patients with pre-existing or recent onset of central nervous system de-myelinating disorders.
Treatment with REMICADE may result in the formation of autoantibodies and, rarely, in the development of a lupus-like syndrome. If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with REMICADE, treatment should be discontinued (see ADVERSE REACTIONS , Autoantibodies/Lupus-like Syndrome ).
Patients with long duration of Crohn' disease or rheumatoid arthritis and chronic exposure to immunosuppressant therapies are more prone to develop lymphomas (see ADVERSE REACTIONS , Malignancies/Lymphoproliferative Disease ). The impact of treatment with REMICADE on these phenomena is unknown.
Treatment with REMICADE can be associated with the development of antibodies to infliximab. One hundred thirty-four of the 199 Crohn' disease patients treated with REMICADE were evaluated for the development of infliximab-specific antibodies; 18 (13%) were antibody-positive (the majority at low titer, <1:20). Patients who were antibody-positive were more likely to experience an infusion reaction (see ADVERSE REACTIONS , Infusion-related Reactions ). Antibody development was lower among rheumatoid arthritis and Crohn' disease patients receiving immunosuppressant therapies such as 6-MP, AZA or MTX. With repeated dosing of REMICADE, serum concentrations of infliximab were higher in rheumatoid arthritis patients who received concomitant MTX. There are limited data available on the development of antibodies to infliximab in patients receiving long-term treatment with REMICADE. Because immunogenicity analyses are product-specific, comparison of antibody rates to those from other products is not appropriate.
No data are available on the response to vaccination or on the secondary transmission of infection by live vaccines in patients receiving anti-TNF therapy. It is recommended that live vaccines not be given concurrently.
Specific drug interaction studies, including interactions with MTX, have not been conducted. The majority of patients in rheumatoid arthritis or Crohn' disease clinical studies received one or more concomitant medications. In rheumatoid arthritis, concomitant medications besides MTX were nonsteroidal anti-inflammatory agents, folic acid, corticosteroids and/or narcotics. Concomitant Crohn' disease medications were antibiotics, antivirals, corticosteroids, 6-MP/AZA and aminosalicylates. Patients with Crohn' disease who received immunosuppressants tended to experience fewer infusion reactions compared to patients on no immunosuppressants (see PRECAUTIONS , Immunogenicity and ADVERSE REACTIONS , Infusion-related Reactions ).
Long-term studies in animals have not been performed to evaluate the carcinogenic potential. No clastogenic or mutagenic effects of infliximab were observed in the in vivo mouse micronucleus test or the Salmonella-Escherichia coli (Ames) assay, respectively. Chromosomal aberrations were not observed in an assay performed using human lymphocytes. Tumorigenicity studies in mice deficient in TNF(alpha) demonstrated no increase in tumors when challenged with known tumor initiators and/or promoters. It is not known whether infliximab can impair fertility in humans. No impairment of fertility was observed in a fertility and general reproduction toxicity study conducted in mice using an analogous antibody that selectively inhibits the functional activity of mouse TNF(alpha).
Since infliximab does not cross-react with TNF(alpha) in species other than humans and chimpanzees, animal reproduction studies have not been conducted with REMICADE. No evidence of maternal toxicity, embryotoxicity or teratogenicity was observed in a developmental toxicity study conducted in mice using an analogous antibody that selectively inhibits the functional activity of mouse TNF(alpha). Doses of 10 to 15 mg/kg in pharmacodynamic animal models with the anti-TNF analogous antibody produced maximal pharmacologic effectiveness. Doses up to 40 mg/kg were shown to produce no adverse effects in animal reproduction studies. It is not known whether REMICADE can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. REMICADE should be given to a pregnant woman only if clearly needed.
It is not known whether infliximab is excreted in human milk or absorbed systemically after ingestion. Because many drugs and immunoglobulins are excreted in human milk, and because of the potential for adverse reactions in nursing infants from REMICADE, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Safety and effectiveness of REMICADE in patients with juvenile rheumatoid arthritis and in pediatric patients with Crohn' disease have not been established.
In the ATTRACT study, no overall differences were observed in effectiveness or safety in 72 patients aged 65 or older compared to younger patients. In Crohn' disease studies, there were insufficient numbers of patients aged 65 and over to determine whether they respond differently from patients aged 18 to 65. Because there is a higher incidence of infections in the elderly population in general, caution should be used in treating the elderly (see ADVERSE REACTIONS , Infections ).
A total of 771 patients were treated with REMICADE in clinical studies. In both rheumatoid arthritis and Crohn' disease studies, approximately 6% of patients discontinued REMICADE because of adverse experiences. The most common reasons for discontinuation of treatment were dyspnea, urticaria and headache. Adverse events have been reported in a higher proportion of patients receiving the 10 mg/kg dose than the 3 mg/kg dose.
An infusion reaction was defined as any adverse event occurring during the infusion or within 1 to 2 hours after the infusion. Nineteen percent of REMICADE-treated patients in all clinical studies experienced an infusion reaction compared to 8% of placebo-treated patients. Among the 4797 REMICADE infusions, 3% were accompanied by nonspecific symptoms such as fever or chills, 1% were accompanied by cardiopulmonary reactions (primarily chest pain, hypotension, hypertension or dyspnea), <1% were accompanied by pruritus, urticaria, or the combined symptoms of pruritus/urticaria and cardiopulmonary reactions. Serious infusion reactions including anaphylaxis were infrequent. Less than 2% of patients discontinued REMICADE because of infusion reactions, and all patients recovered with treatment and/or discontinuation of infusion. REMICADE infusions beyond the initial infusion in rheumatoid arthritis patients were not associated with a higher incidence of reactions.
Patients with Crohn' disease who became positive for antibodies to infliximab were more likely to develop infusion reactions than were those who were negative (36% vs. 11% respectively). Use of concomitant immunosuppressant agents appeared to reduce the frequency of antibodies to infliximab and infusion reactions (see PRECAUTIONS , Immunogenicity and Drug Interactions ).
Reactions following readministration
In a clinical study of forty patients with Crohn' disease retreated with infliximab following a 2 to 4 year period without infliximab treatment, 10 patients experienced adverse events manifesting 3 to 12 days following infusion of which 6 were considered serious. Signs and symptoms included myalgia and/or arthralgia with fever and/or rash, with some patients also experiencing pruritus, facial, hand or lip edema, dysphagia, urticaria, sore throat, and headache. Patients experiencing these adverse events had not experienced infusion-related adverse events associated with their initial infliximab therapy. Of the 40 patients enrolled, these adverse events occurred in 9 of 23 (39%) who had received liquid formulation which is no longer in use and 1 of 17 (6%) who received lyophilized formulation. The clinical data are not adequate to determine if occurrence of these reactions is due to differences in formulation. Patients' signs and symptoms improved substantially or resolved with treatment in all cases. There are insufficient data on the incidence of these events after drug-free intervals of less than 2 years. However, these events have been observed infrequently in clinical studies and post-marketing surveillance at intervals of less than 1 year.
In REMICADE clinical studies, treated infections were reported in 32% of REMICADE-treated patients (average of 37 weeks of follow-up) and in 22% of placebo-treated patients (average of 29 weeks of follow-up). The infections most frequently reported were upper respiratory tract infections (including sinusitis, pharyngitis, and bronchitis) and urinary tract infections. No increased risk of serious infections or sepsis were observed with REMICADE compared to placebo in the ATTRACT study. Among REMICADE-treated patients, these serious infections included pneumonia, cellulitis and sepsis. In the ATTRACT study, one patient died with miliary tuberculosis and one died with disseminated coccidioidomycosis. Other cases of tuberculosis, including disseminated tuberculosis, also have been reported post-marketing. Although the relationship to REMICADE is unknown, most of the cases of tuberculosis occurred within the first two months after initiation of therapy with infliximab and may reflect recrudesence of latent disease (see WARNINGS , RISK OF INFECTIONS ). Twelve percent of patients with fistulizing Crohn' disease developed a new abscess 8 to 16 weeks after the last infusion of REMICADE (see CLINICAL STUDIES , Fistulizing Crohn's Disease ).
In the ATTRACT rheumatoid arthritis study through week 54, 49% of REMICADE-treated patients developed antinuclear antibodies (ANA) between screening and last evaluation, compared to 21% of placebo-treated patients. Anti-dsDNA antibodies developed in approximately 10% of REMICADE-treated patients, compared to none of the placebo-treated patients. No association was seen between REMICADE dose/schedule and development of ANA or anti-dsDNA.
Of Crohn' disease patients treated with REMICADE who were evaluated for antinuclear antibodies (ANA), 34% developed ANA between screening and last evaluation. Anti-dsDNA antibodies developed in approximately 9% of Crohn' disease patients treated with REMICADE. The development of anti-dsDNA antibodies was not related to either the dose or duration of REMICADE treatment. However, baseline therapy with an immunosuppressant in Crohn' disease patients was associated with reduced development of anti-dsDNA antibodies (3% compared to 21% in patients not receiving any immunosuppressant). Crohn' disease patients were approximately 2 times more likely to develop anti-dsDNA antibodies if they were ANA-positive at study entry.
In clinical studies, three patients developed clinical symptoms consistent with a lupus-like syndrome, two with rheumatoid arthritis and one with Crohn' disease. All three patients improved following discontinuation of therapy and appropriate medical treatment. No cases of lupus-like reactions have been observed in up to three years of long-term follow-up (see PRECAUTIONS , Autoimmunity ).
In completed clinical studies of REMICADE for up to 54 weeks, 7 of 771 patients developed 8 new or recurrent malignancies. These were non-Hodgkins B-cell lymphoma, breast cancer, melanoma, squamous, rectal adenocarcinoma and basal cell carcinoma. There are insufficient data to determine whether REMICADE contributed to the development of these malignancies. The observed rates and incidences were similar to those expected for the populations studied 16,17 (see PRECAUTIONS , Malignancy ).
Adverse events occurring at a frequency of at least 5% in all patients treated with REMICADE are shown in Table 4. Patients with Crohn' disease who were treated with REMICADE were more likely than patients with rheumatoid arthritis to experience adverse events associated with gastrointestinal symptoms.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Serious adverse events (all occurred at frequencies <2%) by body system in all patients treated with REMICADE are as follows:
Body as a whole: abdominal hernia, asthenia, chest pain, diaphragmatic hernia, edema, fall, pain
Blood: splenic infarction, splenomegaly
Cardiovascular: hypertension, hypotension, syncope
Central & Peripheral Nervous: encephalopathy, dizziness, headache, spinal stenosis, upper motor neuron lesion
Autoimmunity: lupus erythematosus syndrome, worsening rheumatoid arthritis, rheumatoid nodules
Ear and Hearing: ceruminosis
Eye and Vision: endophthalmitis
Gastrointestinal abdominal pain, appendicitis, Crohn' disease, diarrhea, gastric ulcer, gastrointestinal hemorrhage, intestinal obstruction, intestinal perforation, intestinal stenosis, nausea, pancreatitis, peritonitis, proctalgia, vomiting
Heart Rate and Rhythm: arrhythmia, atrioventricular block, bradycardia, cardiac arrest, palpitation, tachycardia
Liver and Biliary: biliary pain, cholecystitis, cholelithiasis, hepatitis cholestatic
Metabolic and Nutritional: dehydration, pancreatic insufficiency, weight decrease
Musculoskeletal arthralgia, arthritis, back pain, bone fracture, hemarthrosis, intervertebral disk herniation, joint cyst, joint degeneration, myalgia, osteoarthritis, osteoporosis, spondylolisthesis, symphyseolysis, tendon disorder, tendon injury
Myo-, Endo-, Pericardial and Coronary Valve: angina pectoris, cardiac failure, myocardial ischemia
Neoplasms: basal cell, breast, lymphoma, melanoma, rectal adenocarcinoma, skin
Platelet, Bleeding and Clotting: thrombocytopenia
Psychiatric: anxiety, confusion, delirium, depression, somnolence, suicide attempt
Reproductive endometriosis
Resistance Mechanism: abscess, bacterial infection, cellulitis, fever, fungal infection, herpes zoster, infection, inflammation, sepsis
Respiratory: adult respiratory distress syndrome, bronchitis, coughing, dyspnea, pleural effusion, pleurisy, pneumonia, pneumothorax, pulmonary edema, pulmonary infiltration, respiratory insufficiency, upper respiratory tract infection
Skin and Appendages: furunculosis, increased sweating, injection site inflammation, rash, ulceration
Urinary: azotemia, dysuria, hydronephrosis, kidney infarction, pyelonephritis, renal calculus, renal failure, ureteral obstruction
Vascular (Extracardiac): brain infarction, peripheral ischemia, pulmonary embolism, thrombophlebitis deep
White cell and Reticuloendothelial: leukopenia, lymphadenopathy, lymphangitis
A greater proportion of patients enrolled into the ATTRACT study who received REMICADE plus MTX experienced mild, transient elevations (<2 times the upper limit of normal) in AST or ALT (35% and 32% respectively) compared to patients treated with placebo with MTX (24% each). Six (1.8%) patients treated with REMICADE and MTX experienced more prolonged elevations in their ALT.
Single doses up to 20 mg/kg have been administered without any direct toxic effect. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment instituted immediately.
The recommended dose of REMICADE is 3mg/kg given as an intravenous infusion followed with additional similar doses at 2 and 6 weeks after the first infusion then every 8 weeks thereafter. REMICADE should be given in combination with methotrexate. For patients who have an incomplete response, consideration may be given to adjusting the dose up to 10 mg/kg or treating as often as every 4 weeks.
The recommended dose of REMICADE is 5 mg/kg given as a single intravenous infusion for treatment of moderately to severely active Crohn' disease. In patients with fistulizing disease, an initial 5 mg/kg dose should be followed with additional 5 mg/kg doses at 2 and 6 weeks after the first infusion.
There are insufficient safety and efficacy data for the use of REMICADE in Crohn' disease beyond the recommended duration (see WARNINGS , Hypersensitivity ; ADVERSE REACTIONS , Infusion-related Reactions ; and INDICATIONS AND USAGE ).
REMICADE vials do not contain antibacterial preservatives. Therefore, the vials after reconstitution should be used immediately, not re-entered or stored. The diluent to be used for reconstitution is 10 mL of Sterile Water for Injection, USP. The total dose of the reconstituted product must be further diluted to 250 mL with 0.9% Sodium Chloride Injection, USP. The infusion concentration should range between 0.4 mg/mL and 4 mg/mL. The REMICADE infusion should begin within 3 hours of preparation.
Store the lyophilized product under refrigeration at 2°C to 8°C (36°F to 46°F). Do not freeze. Do not use beyond the expiration date. This product contains no preservative.
REMICADE lyophilized concentrate for IV injection is supplied in individually-boxed single-use vials in the following strength:
NDC 57894-030-01 100 mg infliximab in a 20-mL vial
 |
© Centocor, Inc. 2000 License #1242
Malvern, PA 19355, USA Revised 1 December 2000
1-800-457-6399 IN00160